
- HOME
- Custom Synthesis, Peptide Drug (GMP)
- Custom Peptide Synthesis
- Enzyme Substrates and Inhibitors
Enzyme Substrates and Inhibitors
Our company has long been distributing enzyme substrates and enzyme inhibitors, with high reputation from many researchers worldwide. In addition, it is a remarkable trend to synthesize custom substrates and inhibitors specific to the newly discovered enzymes in the various stages from design to synthesis.
Enzyme Substrates (e.g. Protease Substrates)
Enzyme substrate
- wavelength for detection
- Peptide-pNA
- UV 405 nm
- Peptide-MCA(AMC)
- ex. 380 nm / em. 460 nm
- Peptide-AFC
- ex. 400 nm / em. 505 nm, etc.
Fluorescence-Quenching Substrates
- wavelength for detection
- Nma – Dnp
- ex. 340 nm / em. 440 nm
- MOCAc (Mca) – Dnp
- ex. 328 nm / em. 393 nm
- Dabcyl – Edans
- ex. 336-340 nm / em.472-490 nm, etc.
We will also synthesize any fluorescence-quenching compounds in addition to enzyme substrates.
Enzyme Inhibitors (e.g. Protease Inhibitors)
- Peptide Aldehyde (Peptide-CHO)
- Peptide Fluoromethylketone (Peptide-FMK、Peptide-CH2F)
- Peptide Chloromethylketone (Peptide-CMK、Peptide-CH2Cl)
- Boronic Acid (Peptide-B(OH)2)
- Statine Derivatives
- Hydroxymethylcarbonyl isostere
- Epoxide type inhibitors
- Serine Protease Inhibitor (Marinostatin derivatives)
We will design suitable inhibitors for your research. We will also synthesize any compounds in addition to enzyme inhibitors.
Biological active peptides are produced by enzymatic digestions of precursor proteins, which are subsequently degraded and cleared by other enzymes. Research in enzymology is therefore surely critical to clarify the phenomena in the living body. Our company offers two custom services in this field: design and synthesis of A) enzyme substrates and B) enzyme inhibitors.
A) Enzyme Substrates
There are three types of the well-known enzyme substrates: I) chromogenic substrates, II) fluorescent substrates and III) quenching substrates. In the case of I) and II), all the experiments are carried out easily by the aid of the introduced chromophore and fluorophore, respectively. In the case of III), color or fluorescence appears only after enzymatic cleavage of the specific bond between color or fluorescence acceptor and its quencher in the colorless or fluorescentless substrates[1]. It is therefore possible to follow the progress of the enzymatic reaction by measuring the absorbance or fluorescence of the reaction mixture without the chromatographic pretreatment. Typical examples of each type substrate are shown below; we will synthesize any substrates other than those described below depending on your interest. We can participate any and all stages – from design to synthesis – of your enzyme substrates.
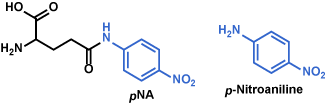
I para-Nitroanilide (pNA) Derivatives (Chromogenic Substrates)
Color of p-nitroaniline generated by enzyme from the p-nitroanilide substrate is yellow, whereas the parental substrate is a mostly white powder. Progress of the enzymatic reaction is monitored by measuring the UV absorbance at 405 nm[2]. Carboxyl group-attached p-nitroaniline is also useful, by which the solubility of p-nitroaniline in water improves[3].
Glu(pNA) [4] (Code 3066)
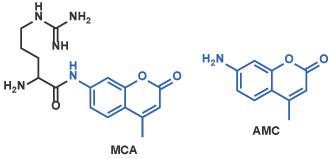
II 4-Methylcoumaryl-7-amide (MCA) Derivatives (Fluorescent Substrates)
Peptidyl-MCA shows the negligible fluorescence. When the enzyme of interest cleaves the bond between the peptide moiety and MCA, strongly fluorescent 7-amino-4-methylcoumarin (AMC) is released[5]; enzymatic reaction is traced by measuring the alteration of fluorescence of the reaction mixture. 7-Amino-4-trifluoromethylcoumarin (AFC) is also used instead of AMC[6].
Arg-MCA [7] (Code 3113-v)
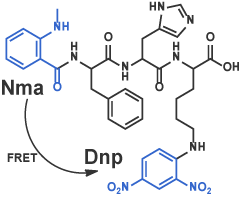
III Fluorescence-Quenching Substrates Based on FRET (Forster Resonance Energy Transfer)
Many fluorescence-quenching substrates, a principle of which is based on FRET, are now known. Once an enzyme cleaves the peptide bond between fluorophore- and fluorescence quencher-attached amino acids, fluorescence of fluorophore recovers by disappearance of FRET effect in proportion to the degree of the enzymatic reaction[8]. In addition to these substrates, more sophisticated substrates are reported frequently; introduction of two fluorophores in substrate and monitoring the enzymatic reaction by detecting the difference in the wavelength of fluorescence afforded by distinct FRET efficacy. We will synthesize any fluorescence-quenching substrates that you designed. Alternatively, we may design a substrate for you by choosing the best combination of fluorophore and fluorescence quencher for the convenience of your experimental purpose.
Nma-Phe-His-Lys(Dnp) [9] (Code 3233-v)
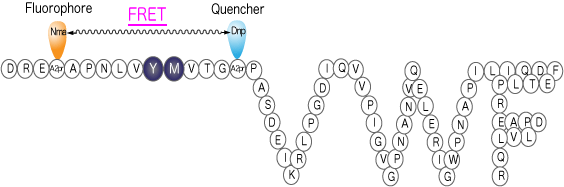
FRETS-VWF73 [10] (Code 3224-s)
B) Enzyme Inhibitors
In the research of enzymology, enzyme inhibitors are as important as enzyme substrates. There are long histories in the development of enzyme inhibitors such as Leupeptin[11], Pepstatin[12], and Chymostatin[13], all of which are discovered in Japan, as well as dipeptidyl peptidase-4 inhibitor[14] for the treatment of type 2 diabetes. Many enzyme inhibitors including those show significant contribution to the quality of human life. Although the recently established method using RNAi is efficient in the inhibition of a specific enzyme, small organic molecule enzyme inhibitors are still indispensable in order to inhibit a particular enzyme directly[15]. In general, enzyme inhibitors are designed by choosing the amino acid sequence near the natural substrate cleavage site in combination with some modifications in the structure. Please find the typical structures of synthetic enzyme inhibitors below, all of which are frequently disclosed in the literature. Do not worry even if you can not find the structure you want to have as an enzyme inhibitor; we will synthesize any structures you designed. If you do not have any special ideas for the structure of an inhibitor against the enzyme of your interest, we can propose the most suitable enzyme inhibitor for your experimental purposes, based on our huge experiences in this field.
<Structure of enzyme inhibitor>
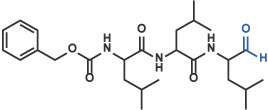
I) Aldehyde
Z-Leu-Leu-Leu-H (aldehyde) [16] (Code 3175-v)
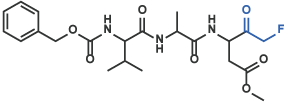
II Fluoromethylketone, Chloromethylketone, Diazomethylketone
Z-Val-Ala-Asp(OMe)-CH2F [17] (Code 3188-v)
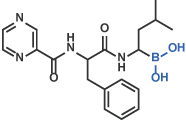
III Boronic acid
Bortezomib [18]
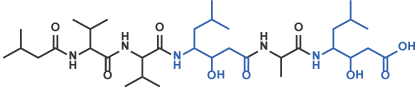
IV Statine derivative
Pepstatin A [12] (Code 4397)
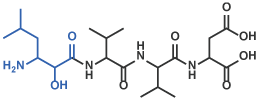
V Hydroxymethylcarbonyl isostere
Amastatin [19] (Code 4095)
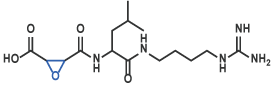
VI Inhibitor including Epoxide
E-64 [20] (Code 4096)
VII Peptidic Serine Protease Inhibitor as scaffold
Among the peptidic and proteinous serine protease inhibitors, chain length of which are in the range between 12 and 190 amino acid residues, marinostatin isolated from a marine organism Pseudoalteromonas sagamiensis is the shortest ever known and is reported to potently inhibit subtilisin. Two side chain ester linkages, one between Thr3 and Asp9 and another between Ser8 and Asp11, are the structural characteristics of this particular inhibitor.
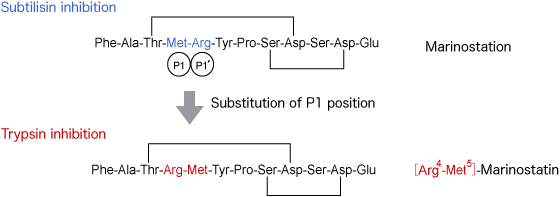
In the structure-activity relationship study of Marinostatin[21], we learned the following fact: substitution of Arg for Met at P1 site converts the subtilisin inhibitor to trypsin inhibitor. Trypsin inhibitory activity of this analog is more potent than that of Leupeptin, implying that Marinostatin may be used as a scaffold for designing an inhibitor to other enzyme. Please contact us if you may be interested in using the constraint structure of Marinostatin for a novel enzyme inhibitor.
- 寺井琢也、長野哲雄 (2011)「第1章 有機蛍光プローブ」菊地和也 編『蛍光イメージング/MRIプローブの開発』シーエムシー出版、p. 1-9。
- B.F. Erlanger, N. Kokowsky, and W. Cohen, Arch. Biochem. Biophys., 95, 271 (1961).
- J.P. Persijn, and W. van der Slik, J. Clin. Chem. Clin. Biochem., 14, 421 (1976).
- M. Orlowski, and A. Meister, Biochim. Biophys. Acta., 73, 679 (1963).
- M. Zimmerman, E. Yurewicz, and G. Patel, Anal. Biochem., 70, 258 (1976).
- R.E. Smith, E.R. Bissell, A.R. Mitchell, and K.W. Pearson, Thromb. Res., 17, 393 (1980).
- Y. Kanaoka, T. Takahashi, H. Nakayama, K. Takada, T, Kimura, and S. Sakakibara, Chem. Pharm. Bull., 25, 3126 (1977).
- S.A. Latt, D.S. Auld, and B.L. Vallee, Anal. Biochem., 50, 56 (1972).
- S. Takahashi, H. Ono, T. Gotoh, K. Yoshizawa-Kumagaye, and T. Sugiyama, Biomed. Res., 32, 407 (2011).
- K. Kokame, Y. Nobe, Y. Kokubo, A. Okayama, and T. Miyata, Br. J. Haematol., 129, 93 (2005).
- S. Kondo, K. Kawamura, J. Iwanaga, M. Hamada, T. Aoyagi, K. Maeda, T. Takeuchi, and H. Umezawa, Chem. Pharm. Bull., 17, 1896 (1969).
- H. Umezawa, T. Aoyagi, H. Morishima, M. Matsuzaki, M. Hamada, and T. Takeuchi, J. Antibiot., 23, 259 (1970).
- H. Umezawa, T. Aoyagi, H. Morishima, S. Kunimoto, M. Matsuzaki, M. Hamada, and T. Takeuchi, J. Antibiot., 23, 425 (1970).
- C. F. Deacon, Diabetes, Obesity and Metabolism, 13, 7 (2011).
- (a) U. S. Eggert, C. M. Field, and T. J. Mitchison, Mol. BioSyst., 2, 93 (2006) (b) W. A Weiss, S. S Taylor, and K. M.Shokat, Nat. Chem. Biol., 3, 739 (2007).
- Y. Saito, S. Tsubuki, H. Ito, and S. Kawashima, Neurosci. Lett., 120, 1 (1990).
- H. Zhu, H.O. Fearnhead, and G.M. Cohen, FEBS Lett., 374, 303 (1995).
- J. Adams, Cancer Cell, 5, 417 (2004).
- T. Aoyagi, H. Tobe, F. Kojima, M. Hamada, T. Takeuchi, and H. Umezawa, J. Antibiotics., 31, 636 (1978).
- K. Hanada, M. Tamai, M. Yamagishi, S. Ohmura, J. Sawada, and I. Tanaka, Agric. Biol. Chem., 42, 523 (1978).
- M. Taichi, T. Yamazaki, K. Kawahara, D. Motooka, S. Nakamura, S. Harada, T. Teshima, T. Ohkubo, Y. Kobayashi, and Y. Nishiuchi, J.Pept.Sci., 16, 329 (2010).